DISEASE &
TREATMENTS
Click to know more about neurological disorders and diseases and it's treatment.
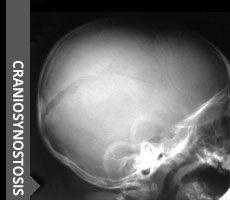
Skull deformities are a very common reason for referral to pediatric neurosurgery.Premature fusion of cranial suture closure is called craniosynostosis.
Incidence and Aetiology :
Isolated craniosynsostosis occurs due to the premature fusion of a single suture or multiple sutures. The overall prevalence of craniosynostosis is around 1 in 2500 live births, with ranges of 1 in 709 to 1 in 3225 of births. The wide variation seen is due to differences in the various criteria under which one labels craniosynsostosis. Isolated craniosynostosis is far more common than multiple ‘syndromic’ synostosis.Sagittal seems to be commonest, followed by coronal and metopic. True lambdoid synostosis appears rare and is difficult to distinguish from deformational posterior asymmetry, which is considered to be because of positional molding of the cranium.
Common Syndromic Craniosynostosis like Apert and Crouzon have prevalence of about 1 in 65000, each about 4-6 per cent of all craniosynostosis. There is evidence of FGFR2 mutation in Apert Syndrome, FGFR3 in Crouzon syndrome and FGFR1 in Pfeiffer syndrome. Almost all Aperts and about 50 per cent of Crouzons arise de novo and there is a correlation with increase in paternal age. The prevalence of Pfeiffer syndrome is lower than others but there is no definite percentage data.
Apart from this there are rarer types of single gene syndromes associated with other malformations, metabolic storage disorders and learning disabilities.There are chromosomal aberrations of which craniosynostosis may be a feature. There are teratogenic causes associated with anticonvulsants, (Valproate, Hydantoin)cytotoxic agents, (Methotraxate, Cyclophosphomide, Cytarabine) abortifacients, and nitrosatable drugs(Chlorpneramine,Nitrofurantoin, Chlordiazepoxide and Fluconazole).
Classification and Terminology
Over the years, the nomenclature for various craniofacial deformities has remained constant, but with the entry of molecular genetics, there have been additional terms given to the complex multi sutural synostosis. However, in simple terms these deformities can be classified into three different varieties.
Complex craniofacial Deformities
These are mainly associated with syndromes and FGFR mutations and they are named as follows
Crouzon syndrome was described by Crouzon in 1912. Apart from craniosynostosis this condition manifests maxillary hypoplasia, exorbitsm with hyperteleorism, and typeIII malocclusion. It has no obvious skeletal anomalies. There may associated FGFR3 mutation and phenotypic variability between generations is a common observation.
Apert syndrome was described by Apert in 1906. It is characterized by bicoronal and anterior Sagittal synostosis, midface retrusion and hypertelorism with orbital proptosis and complex syndactyly of hands and feet. They can also have sporadic visceral malformations and skin abnormalities.
Saethre-Chotzen Syndrome is so named after the two people who described their findings in different families around 1931-32. The features here include Bicoronal synostosis with variable involvement of the Metopic, Sagittal and Lambdoid sutures. There is associated proptosis, low set frontal hairline, parrot beaked nose, strabismus, and brachydactyly.
Pfeiffer Syndrome has features of bicoronal synostosis to pan-sutural synostosis and clover leaf deformity of the skull. It was described by Pfeiffer in 1964 and it includes limb anomalies like thick broad thumb and toe, fusion between thumb phalanges, and soft tissue syndactyly of thumbs and fingers.
Clover Leaf or Kleeblattschadel Deformity is an uncommon and severe form of craniosynostosis resulting from the fusion of both coronal and a combination of metopic, lambdoid and sagittal sutures. The midface is retruded and is almost ocular ectopia and dental malposition. The posterior fossa is small and there is cerebellar tonsilar herniation. There may be severe airway obstruction and the clinical prognosis is often poor, unless improved by some very aggressive surgical management.
Carpenter syndrome is a rarer type of syndrome where the cranial deformity mainly involves Sagittal and lambdoid sutures asymmetrically and severely. The affected person is usually short, obese and neuro-developmentally retarded.
Antley-Bixler syndrome is also a rare type of synostosis where there is involvement of coronal and lambdoid sutures, with dysplastic ears, radiohumeral synostosis, long tapering fingers and upper airway abnormalities.
Secondary Craniosynostosis
These can occuras a result of twinning, amniotic bands, or uterine abnormalities and is seenin preterm babies. It can also occur in newborns with hyperthyroidism ormaternal hyperthyroidism during pregnancy.(Hirano et al 1995, Gardener et al1998). Pan sutural synostosis is seen in children with microcephaly where thebrain is small and under developed.
Imaging
Any unusual head shape at birth usually sets off a series of investigations which include Skull XRays, CTScan of head, MRI and Xrays of hands, feet and the cervical spine.
Plain XRay of the Skull is the most used modality and is a good investigation to assess the primary condition and to confirm that there is no other contributory factor like dysplasia or just positional change in shape.Skull Xray AP and AP Townes with Lateral view is capable of showing sutural absence, para sutural sclerosis, sutural narrowing and heaping or enostosis and also to confirm whether the entire suture or only part of it is involved.
CTScan of the head and brain, with 3-D reconstruction, gives an even better picture of the skull bones and sutural fusion, along with the deformity of the sphenoid plate and the base of skull asymmetry.
MRI scan is helpful in syndromic children to define the airways, the craniovertebral junction anomalies, hydrocephalus, venous sinus anatomy and any other cerebral or cerebellar anomalies as also upper laryngo-pharyngeal airway. Hydrocephalus may occasionally be associated with the syndromic synostosis like Apert, Crouzon or Clover-leaf syndrome, and be picked up during investigations. If decompression is carried out early, then hydrocephalus may regress, and if not, may require a shunt surgery. The mechanism of hydrocephalus may be aqueductal stenosis, or venous drainage obstruction in a crowded posterior fossa.
Treatment for craniosynostosis
The treatment of children with this deformity is a challenging surgical task. As mentioned earlier it requires a close coordination with specialists in Neurosurgery, Plastic Surgery, Maxillo-Facial surgery, Orthodontics, Ophthalmologists,
Depending on the severity of the condition and its pressure on the brain surgery is warranted. Most cases indication for surgery is cosmetic reasons however certain cases of Craniosynostosis when more than one suture has fused, surgery is done to prevent raised pressure in the brain. All children receive blood transfusion, Auto-absorbing mini-plates are used during surgery, which absorb in nine months to prevent patients from having long-term hardware implants.. Patients stay in the PICU for couple of days and generally are discharged in three to five days
Total cranial vault reconstruction. The concept is now the accepted surgical treatment of craniosynostosis .We as a part ofcraniofacial team holds a monthly craniofacial clinic and includes specialists from Pediatric Neurosurgery, Craniofacial plastic surgery, Neuroradiology, Neuro-ophthalmology,and speech therapy , Early referral is preferred because the ideal timing for surgical intervention of most Craniosynostosis is at six months of age. The precise pathology and treatment plan is discussed with the family in detail and all patients undergo preoperative CT scanning.
The most common reason for referral is positional plagiocephaly also known as benign positional moulding. Most cases of positional plagiocephaly are treated conservatively.
Click to know more about neurological disorders and diseases and it's treatment.

© 2016 ChildNeuroSurgeon.com. All rights reserved | Design by MHI